2020/05/14 インストールするプログラム等の解説を追記
2021/05/21 Trinity, gcc を最新版に変更、Apple Silicon (M1) Mac を使う場合の設定を追記
2022/12/10 記述内容を修正。初学者に解りにくい部分を改善(したつもり)。
解析環境の構築
このページでは、Trinityを使ってアセンブリーや発現量解析を行うための環境構築に特化して説明しているので、一般的な用途での解析環境とは異なる。Anaconda 標準の解析環境だと、Trinity で必要になる各種パッケージをインストールする際に、ソフトウエア間の依存関係の調査にめちゃくちゃ時間がかかるようになったり、競合によってめちゃくちゃ古いバージョンしかインストールできなくなったりするため。
データ解析など、研究の現場ではキーボードからコマンドを入力して操作するソフトウエアが多く使われる。macOSはUNIXをベースとしているので、コマンドライン操作が行えるターミナルアプリが標準で付属するため、解析環境を構築するためのハードルが低いのでお薦めである。
このマニュアルでは、コマンド入力をするところという意味で「:>」という記号を行頭につける。この記号は除いて、後のコマンドを改行せずに一行で入力する。
Macの選び方
Mac miniやiMacといった一般向けの機種で計算性能は十分ではあるが、メモリーは多い方が良い。MacBook Proなどノート型でも大抵の解析作業は行えるが、計算負荷が大きくなると冷却ファンの音が煩くなる可能性があるので、デスクトップ型が良いのではないかと思う。個人的には、最大限のメモリーを積んだMac miniがお薦めである。
Apple Silicon (M1・M2) Mac を使用してトラブルが起きる場合は、
アプリケーション > ユーティリティ > ターミナル
を選択して
ファイル > 情報を見る
を表示、「Rosettaを使用して開く」にチェックを入れる。
これにより、Apple Silicon に未対応のコマンドやツール群も使用可能になり、解析に関わる大多数のトラブルが解消できる。
(2022年以降は多くのソフトウエアがApple Siliconにネイティブ対応しているので、CPUアーキテクチャが原因のトラブルはほぼなくなった)
必要なソフトウエアをインストールする
- MacPorts
ソフトウエアパッケージ管理環境のひとつ。何かソフトをインストールする際、依存関係がある別のソフトがある場合には一緒にインストールしてくれるなど、環境構築や管理を便利にしてくれる。同様の管理システムに Fink や HomeBrew があるが、個人的に MacPorts が最も'行儀が良い'と感じている。特にAnacondaと混在させる場合には最良の選択肢だと思われる。
- Trinity
RNA-seqのリードデータから、転写産物の塩基配列を構築(アセンブル)するためのソフト。付属するツール群が優秀なので、Macの性能的にアセンブルが不可能であったとしても、解析環境として手元で動かせるのはメリットが大きい。
- Anaconda
Python + データ解析パッケージ の環境を構築するのに人気の高い管理システム。最近はPythonの解析パッケージだけではなく、バイオインフォマティクスの各種ツールがAnacondaの中のBiocondaというチャンネル内で配布されており、生物学に関係する人には欠かせないシステム。
- R
統計解析に特化したプログラミング言語。これも研究者には欠かせないシステムの一つで、トランスクリプトーム解析でも多用する。Rの環境を構築する方法は色々あるが、使用するパッケージをインストールしようとしてもコンパイルが通らないといったトラブルが起きることもあり、管理はやや面倒。Anacondaを使うとRのパッケージも簡単にインストールできるので楽チン。Trinity + 発現量解析を行うという前提では、Anaconda内で環境構築することをお勧めする。
以下、順を追って解説する。
0 念のため、シェルを確認する
:> echo $SHELL
と入力して
/bin/zsh と出ればOK
違う場合は :> chsh -s /bin/zsh
シェルに好みがある場合はそれを使っても構わないが、ここではzshを使う前提で解説を行う。適宜、.zshrc を適切な環境設定ファイルに読み替える。例えば、bash を使いたい場合は .zshrc を .bashrc に変更して下記の作業を行う。
用語解説
シェル: コマンドラインでコンピューターを操作する際、人間とコンピューターの間を取り持つ役割の環境プログラム。tcsh, bash, zsh などがある。POSIX準拠のUNIXではtcsh、Linuxではbashが標準で使われることが多い。macOSでは、以前はbashが標準であったが、Catalina以降はzshが標準になった。どれを使ってもできることはほぼ一緒で、ちょっとした方言の違い程度なので好みのものを使えば良い。特に希望がなければ、macOS標準のzshにしておくのが無難である。
コンパイル: プログラムのソースコードから、ソフトウエアの実行形態に変換すること。
Mac App Store から CotEditor (無料のコード・テキストエディター)をインストール
(必須ではないが、コード作成等で便利なのでオススメ)
使い方 (環境設定ファイルを編集する場合)
:> open -a coteditor .zshrc
1 Xcode コマンドラインツールをインストール
Terminal.app を開いて、下記のコマンドを入力する。
:> xcode-select --install
2 MacPorts をインストール
UNIX系のソフトウエアパッケージ管理環境
https://www.macports.org/install.php
パッケージ管理環境にはいくつかあって、おそらくMac用としては HomeBrew が最も利用者多い。利用者が多い分、Google検索等で必要な情報を得やすいというメリットがあるものの、HomeBrew は Anaconda との相性に問題が生じることが多い。バイオインフォマティクスで利用するソフトウエアは、Anaconda のチャンネルの一つである Bioconda で管理されているものが多くて便利なので、Anaconda の使用は外せない。そこで、Anaconda との相性問題が生じない MacPorts をパッケージ管理環境として利用する。MacPorts のもう一つのメリットとして、Mac 用の Xwindow システムである Xorg のメンテナンスが継続されているので、長期的に見て安心感がある。
とは言っても、MacPorts を使ってインストールするものは、必要最小限になるのであまり使わない。
インストール完了後、
:> sudo port selfupdate
と入力して
The ports tree has been updated. To upgrade your installed ports, you should run port upgrade outdated
と表示されたらOK。
Trinityのコンパイルに必要なソフトを導入する。
最初にgccコンパイラをインストールする。このマニュアルの更新が追いついていない場合があるので、下記のバージョン表記にはこだわらずに導入時点の最新版を使うと良い。
:> port search gcc
で様々なバージョンが表示される。マニュアル更新時点では gcc12 が最新。
:> sudo port install gcc12 cmake
ついでに、ファイルの加工に使うソフト等もインストール
:> sudo port install gsed xorg-server
3 Trinity をコンパイルする
後の手順でインストールするAnacondaの環境が、Trinityのコンパイルの阻害になるので先にこちらを行う。
※既に Anaconda 環境を構築して利用している場合は、一時的に無効化する必要がある。下記「おまけ1」を参照。
MacPorts の gcc が使われるようにセットする
:> port select --list gcc (インストールされたgccを確認)
:> sudo port select gcc mp-gcc12
Trinityの最新版のソースコードをダウンロードする。
https://github.com/trinityrnaseq/trinityrnaseq/releasesホームディレクトリーなど、分かりやすいところにファイルを置いて、展開する。展開したフォルダーの名前もわかりやすいものに変更する。
例: Analysis/Trinity-v2.12.0
ソースコードから実行形式にコンパイルする。
(次の項目のAnacondaをインストールする前に行う)
:> cd ~/Analysis/Trinity-v2.12.0
:> make && make plugins
Trinity のソースコードよりも新しいバージョンのGCCコンパイラが使われている場合、autoconfのバージョンが合わないといったエラーが出ることがある。このようなエラーメッセージがでてコンパイルが止まった場合、MacPorts で適切なバージョンに入れ替えると解決する。
終了したら、Trinityの場所を設定する。
:> echo 'export TRINITY_HOME=$HOME/Analysis/Trinity-v2.12.0' >> ~/.zshrc
起動するか確認する。
:> $TRINITY_HOME/Trinity
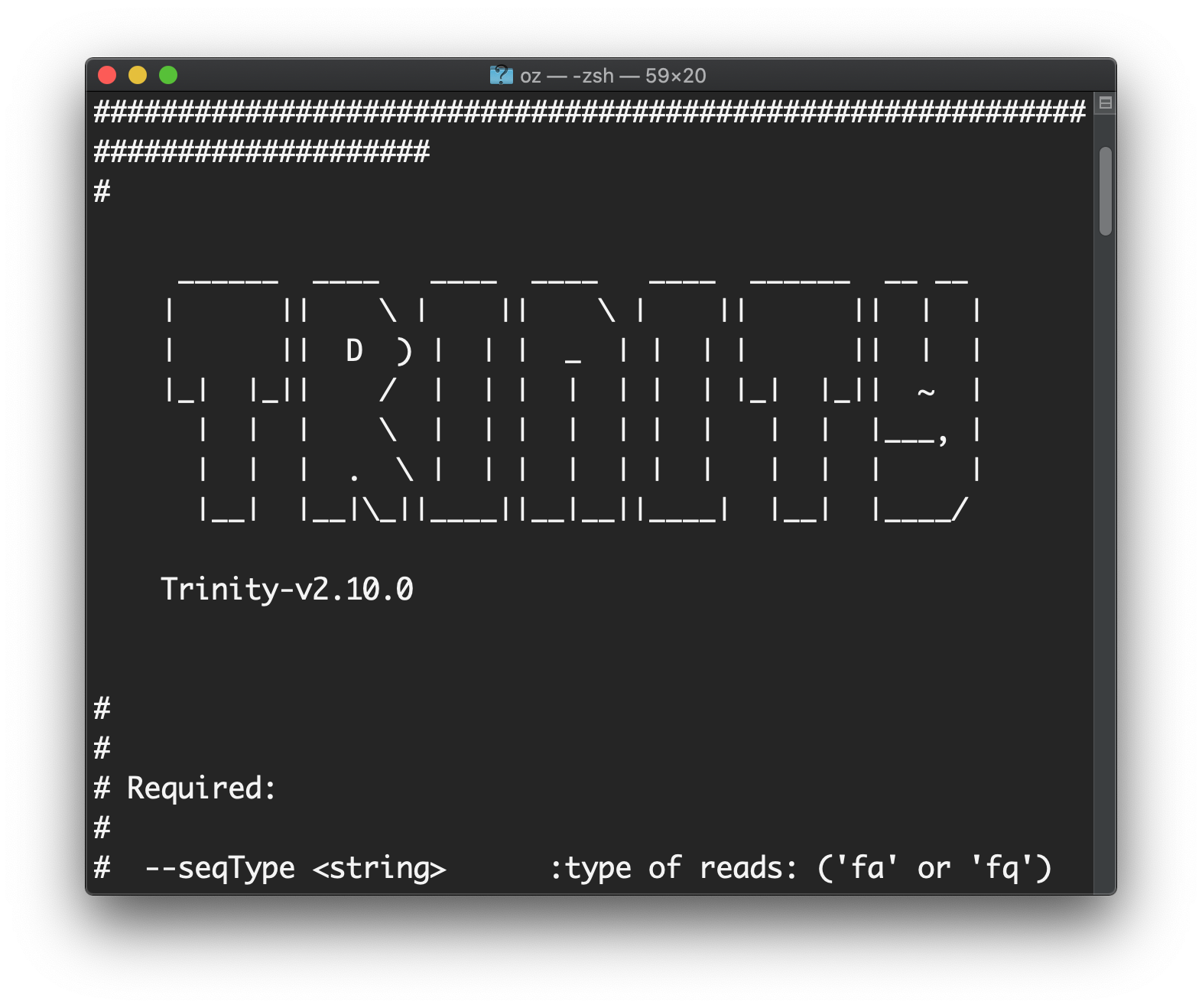
こんな画面が出たらOK!
gcc を標準のものに戻す
:> sudo port select gcc none
4 Anaconda をインストール
公式サイトからダウンローダーを入手して、通常の操作でインストール。
Apple Silicon Mac を使っている場合も、x86_64版をダウンロードする。arm64版だと、Rなど必要なソフトウエアに利用できないものが多くて、環境構築が難しい。
https://www.anaconda.com/distribution/
グラフィカルユーザーインターフェースが必要ない場合、最小構成の Miniconda をインストールしても良い。その後の conda コマンドの利用については同じなので、ほとんど困ることはない。
ターミナルでAnaconda にチャンネルを追加する(下記の順番を守ること)
:> conda config --append channels bioconda
:> conda config --append channels anaconda
:> conda config --append channels conda-forge
この順番でチャンネルを追加すると、デフォルトのチャンネルを優先しつつ、必要なときには追加したチャンネルも使うようになる。インストールされた関連ファイルのバージョンが新しすぎることによるトラブルを回避できるので、この設定がオススメ。
Python 3.10 環境を作る(この段階ではRはインストールしない)
Anaconda Navigator を起動する
Envronments で「Create」をクリック
py310 など、わかりやすい短い名前をつけて新規環境を作成
python のリストから 3.10 を選択
r のチェックは外したまま
:> echo 'conda activate py310' >> .zshrc
ターミナルの新しいウインドーを立ち上げる
この状態では、Pythonはインストールされているもののそのほかはない最小限の環境になる。
必要なソフトのインストール
各ソフトの最新版は、searchコマンドで確認できる。
例
:> conda search bowtie
- bowtie 1.3 以上
- bowtie2 2.5 以上
- kmer-jellyfish 2.3 以上
- salmon 1.9 以上
- samtools 1.16 以上
- blat 35 以上
例: インストール可能な最新版を指定する
:> conda install bowtie=1.3
ソフト名をスペースでつなげて、まとめてインストールすることもできる。このマニュアルの手順に従ってインストールすれば、ほとんどが最新版か最新に近いバージョンになるはず。インストールについて確認されるので、念のためバージョンが上記以上であるかチェックする。
:> conda install salmon kmer-jellyfish bowtie2 samtools blat
最新版かどうか気になる場合は、
:> conda search パッケージ名
として最新版を確認して、バージョンを指定してインストールする。
発現量解析で必要になるソフトをインストールする
最初にBlast
:> conda install blast=2.12(その時点での最新版)
この他に必要なもの
- biopython=1.78
- eXpress=1.5
- fastqc=0.11
- kallisto=0.48
- seqkit=2.3
- transdecoder=5.5
- trimmomatic=0.39
- trinotate=3.2
まとめてインストール
:> conda install kallisto eXpress trimmomatic transdecoder seqkit fastqc trinotate biopython
5 Rに必要なパッケージをインストール
RSEMの発現量解析などで使うパッケージ。これはR内でインストールするとコンパイルエラーが出たり何かと面倒なのだが、conda でインストールすると簡単に終わる。
ただし、Anaconda の環境に conda で管理するRを追加すると、ターミナルの起動が遅くなってしまうので若干使い勝手に影響する。Trinity + 発現量解析に特化した環境として割り切る。
Rを公式サイトや別のパッケージ管理システムを使って既にインストールしている場合は、そちらを使う方が良い場合もある。
:> conda install r-base=4(R4.2系はSalmonとコンフリクトするので、4.1系がインストールされればOK)
:> conda install rsem(1.3以上)
:> conda install r-gplots r-ape r-argparse r-fastcluster
:> conda install bioconductor-seqlogo bioconductor-edger bioconductor-deseq2 bioconductor-ctc bioconductor-biobase bioconductor-qvalue bioconductor-goseq
ターミナルで「R」を実行し、主要なライブラリーのロードが問題なく実行できることを確認する
:> R
:> library(edgeR)
:> library(DESeq2)
:> q()
以後、R内で何かを実行して「○○が足りない」というエラーが出て止まった場合、その名前で conda を検索してヒットしたものをインストールすると解決する。
例
library(goseq) でエラー:
‘goseq’ という名前のパッケージはありません
実行が停止されました
:> conda search goseq
:> conda install bioconductor-goseq
6 テストアセンブルを実行1 簡易版
:> cd $TRINITY_HOME/sample_data/test_Trinity_Assembly
:> ./runMe.sh
7 おまけ1
新しいバージョンのTrinityが出たり、解析環境構築後にTrinityのコンパイルを行う必要があるときは、Anacondaの環境を無効化する必要がある。そのためには、Anacondaのインストーラーがシェルの環境設定ファイルに書き込んだ設定部分を、一時的に削除(またはコメントアウト)する。
.zshrc を開いて
# >>> conda initialize >>>
# !! Contents within this block are managed by 'conda init' !!
__conda_setup="$('/Users/名前/opt/anaconda3/bin/conda' 'shell.zsh' 'hook' 2> /dev/null)"
if [ $? -eq 0 ]; then
eval "$__conda_setup"
else
if [ -f "/Users/名前/opt/anaconda3/etc/profile.d/conda.sh" ]; then
. "/Users/名前/opt/anaconda3/etc/profile.d/conda.sh"
else
export PATH="/Users/名前/opt/anaconda3/bin:$PATH"
fi
fi
unset __conda_setup
# <<< conda initialize <<<
conda activate py38
の部分を削除(またはコメントアウト)して保存し、新しいターミナルのウインドーを開いてTrinityをコンパイルする。
コンパイルが完了したら.zshrcを元に戻してAnacondaが使える状態にし、新しいターミナルのウインドーを開いてcondaコマンドを動かしてみるなど動作確認を行う。
8 おまけ2
発現量解析で使うRの統計解析アルゴリズム「DESeq2」を読み込むとき、下記のエラーが出る場合がある(2020/05/01現在)。
共有ライブラリ '/Users/名前/opt/anaconda3/envs/py38/lib/R/library/DESeq2/libs/DESeq2.dylib' を読み込めません:
dlopen(/Users/名前/opt/anaconda3/envs/py38/lib/R/library/DESeq2/libs/DESeq2.dylib, 6): Library not loaded: @rpath/libopenblasp-r0.3.7.dylib
Referenced from: /Users/名前/opt/anaconda3/envs/py38/lib/R/library/DESeq2/libs/DESeq2.dylib
これはDESeq2が必要とする openblas のライブラリーがインストールされてはいるものの、読み込もうとしている version 0.3.7 より新しいものになっている場合に起きる。openblasのバージョンを下げれば解決するので、
:> conda install openblas=0.3.7
とバージョンを指定してインストールし直すことで解決する。Anaconda で管理するソフトウエアやパッケージは、依存関係があるソフトを一緒にインストールしてくれるものの、バージョンが合わないということが時々あるので、この方法を覚えておくと解決が早くなる可能性がある。
目次
- 環境構築
- リードのクオリティチェックとアセンブル
- Transdecoder による遺伝子予測とアノテーション
- リード数のカウント
- カウントマトリクスの作成
- リードカウントのQC解析
- DE解析
- DEG取り出し
- おまけ1: Transdecoderの自動化
- おまけ2: 発現量解析の自動化
- おまけ3: Anacondaコマンドの使い方一覧
Q&A
皆様からいただいた質問と回答など、こちらへ掲載します。-
先にAnacondaがインストールされていた場合
- テストアセンブルがエラーで停止する
- Apple Silicon (M1, M2) Mac で Trinity のコンパイル途中にエラーが出て止まる場合